General info
Trial chair: David Cibula
CEEGOG number: CEEGOG CX-05
ENGOT number: ENGOT-cx16/CEEGOG/CERVANTES
ClinicalTrials.gov number: NCT04989647
EU CT number: 2023-504973-19-01
Overview
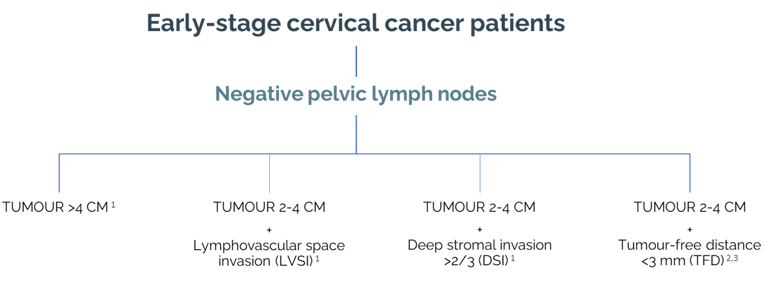
The role of adjuvant therapy in intermediate risk (IR) cervical cancer patients is controversial, supported by a single randomised study performed more than 20 years ago (GOG 92 study, 1999). The intermediate-risk group is defined as lymph-node-negative but with a combination negative prognostic factors (tumour size >2 cm, lymphovascular space invasion, deep stromal invasion >2/3). Recent retrospective studies showed excellent local control in intermediate-risk group patients after radical surgery with no additional adjuvant treatment.
The CERVANTES trial is designed to bring level A evidence on the role of adjuvant therapy in IR patients in an international, prospective, randomised study. Patients will be registered and randomised after radical surgery if the final pathology report has confirmed IR group and other inclusion criteria into ARM A, with no additional treatment, and ARM B, receiving adjuvant therapy. The quality assurance program will be in place for adjuvant external beam radiotherapy. Accrual is expected to last for 5 years, with an additional 3 years of follow-up for primary endpoint analysis. If the primary analysis shows a positive outcome, the study will continue for 3 further years to evaluate overall survival as a secondary endpoint.
The objective of the trial is to evaluate if adjuvant therapy is associated with disease-free survival benefit after radical surgery in patients with IR cervical cancer. The primary endpoint of the study is disease-free survival from the day of randomisation. A total of 514 patients (including an expected drop-out rate of 10%) are required to achieve 80% power on 5% significance level with non-inferiority margin of 5% to test the difference between the arms using the Cox proportional hazards model. The maximal tolerated margin for non-inferiority in 2-year DFS is 5%.
Intermediate-risk (IR) group
Arms
and
interventions


CERVANTES
Arm
Interventions/treatment
Arm A: Surgery only
Arm A:
IR cancer patient underwent surgery consisting of radical hysterectomy, sentinel lymph node (SLN) biopsy and systemic pelvic lymphadenectomy (PLND)*. No further treatment will be administered
*PLND could be avoided in patients with tumours <4 cm
Patient will not receive any type of adjuvant therapy
Arm B: Surgery + adjuvant therapy
IR cancer patient undergone surgery consisting of radical hysterectomy, sentinel lymph node (SLN) biopsy and systemic pelvic lymphadenectomy (PLND)*, followed by adjuvant treatment
*PLND could be avoided in patients with tumours <4 cm
Arm B:
Patient will receive adjuvant treatment composed of pelvic external radiotherapy (EBRT) ± brachytherapy ± concomitant chemotherapy
Outcome measures
CERVANTES
Primary endpoint
Key secondary endpoint
Disease-free survival
Overall survival
Calculated as an interval from the day of randomisation until diagnosis of recurrence:
unequivocal finding on imaging by subjective radiological assessment
suspicious recurrence on imaging either confirmed by biopsy or supported by other signs (disease progression on imaging or progression of symptoms)
physical examination supported by clinical evidence (i.e., symptoms or progression)
death caused by disease or death of unknown cause
In case of a positive outcome of the primary DFS analysis, the trial will continue for a further 3 years to evaluate overall survival
Other secondary endpoints
Pelvic disease-free survival
Health-related quality of life
Treatment-related adverse events
Subscribe for CERVANTES Trial
newsletter*